

Computational chemistry, the branch of chemistry that employs computers and algorithms both for the prediction of molecular properties and the simulation of molecular behavior, has a long history of utilizing all of the power made available from state-of-the-art computing.
Soon after the foundations of quantum mechanics were established in the early 1920’s, researchers began applying these new theoretical tools to the study of single atoms and the very simplest molecules in attempts to understand their behavior. Very early in the development of computational chemistry was the realization that even the fastest available computers were insufficient to handle the mathematical complexity needed to obtain “chemical accuracy,” the prediction of molecular properties as observed by experimentalists. As a result, computational chemistry progressed along two lines – the development of codes to perform the calculations and the development of approaches to reduce the computational workload through simplification of the mathematical descriptions of nuclei and electrons. In some cases, the developed approximations were used to simplify the description of electrons in their orbitals, including simplifying the models used to describe how multiple electrons interact with one another in chemical bonds. In other cases, the ability to model chemical bonding was deemed less important than the ability to model the ways in which stable molecules interact with one another through structural and electrostatic changes – this route lead to the abandonment of quantum theory in favor of less rigorous Newtonian mechanics descriptions still used broadly in the pharmaceutical industry.
Only in the last few decades have computational resources become both available and cost-effective for the high-level study of chemical behavior. Computational chemistry has developed to a point where researchers can now obtain meaningful predictions from advanced quantum chemical studies in nearly the time it takes to perform the associated chemical studies on the molecules themselves in the laboratory – a considerable testament to the power of modern computing hardware and the application of modern algorithms to adequately approximate chemical reality. With the introduction of campus-wide virtual computing networks that interconnect universities around the world, computational chemists have seen rapid growth in both the development and validation of theoretical methods, all driven by the ability to generate massive amounts of data by which to gauge the accuracy of models and algorithms that often date back decades.
Common Tools Support a Variety Of Departmental Projects
The three broad areas of research in Computational Chemistry – Quantum Chemistry, Molecular Dynamics, and Informatics – are all represented on OrangeGrid, Syracuse University’s High Throughput Computing (HTC) environment. Within the Department of Chemistry alone, quantum chemistry tools have been used to study a diverse array of projects such as the coordination chemistry of alkali and alkali earth metal complexes (K. Ruhlandt), the binding of bioconjugates to transport proteins for therapeutic applications (R. Doyle), nanoscale self-assembly on boron nitride thin films and the electronic properties of aromatic molecular clusters (J. Spencer), molecular crystal structure formation and molecular electronics (B. Hudson), and the development of new computational tools to support the study of functional molecules and nanoscale materials (A. Chakraborty).
One area of computational chemistry that has seen considerable development in the last decade is the application of quantum chemical tools to the study of molecular solids. While the underlying mathematics of solid-state chemistry have existed for decades, the study of molecular solids is particularly challenging – the high-level descriptions of electrons and their behavior are multiplied by the need to account for the interactions between molecules in their highly-ordered, periodic crystal environments.
When Significant Resource Needs Produce Significant Results
Dr. Timothy Korter, Professor and Chair of the Department of Chemistry, was among the first to demonstrate that the application of solid-state computational chemistry for the study of molecular behavior is a necessity for correctly interpreting experimental data. The Korter Group is highly regarded for their contributions to the field of Terahertz (THz) Spectroscopy – a branch of Physical Chemistry that exploits the low-frequency infrared spectrum for the study of large-scale motions of molecules and the motions between molecules in liquid and solid phases.
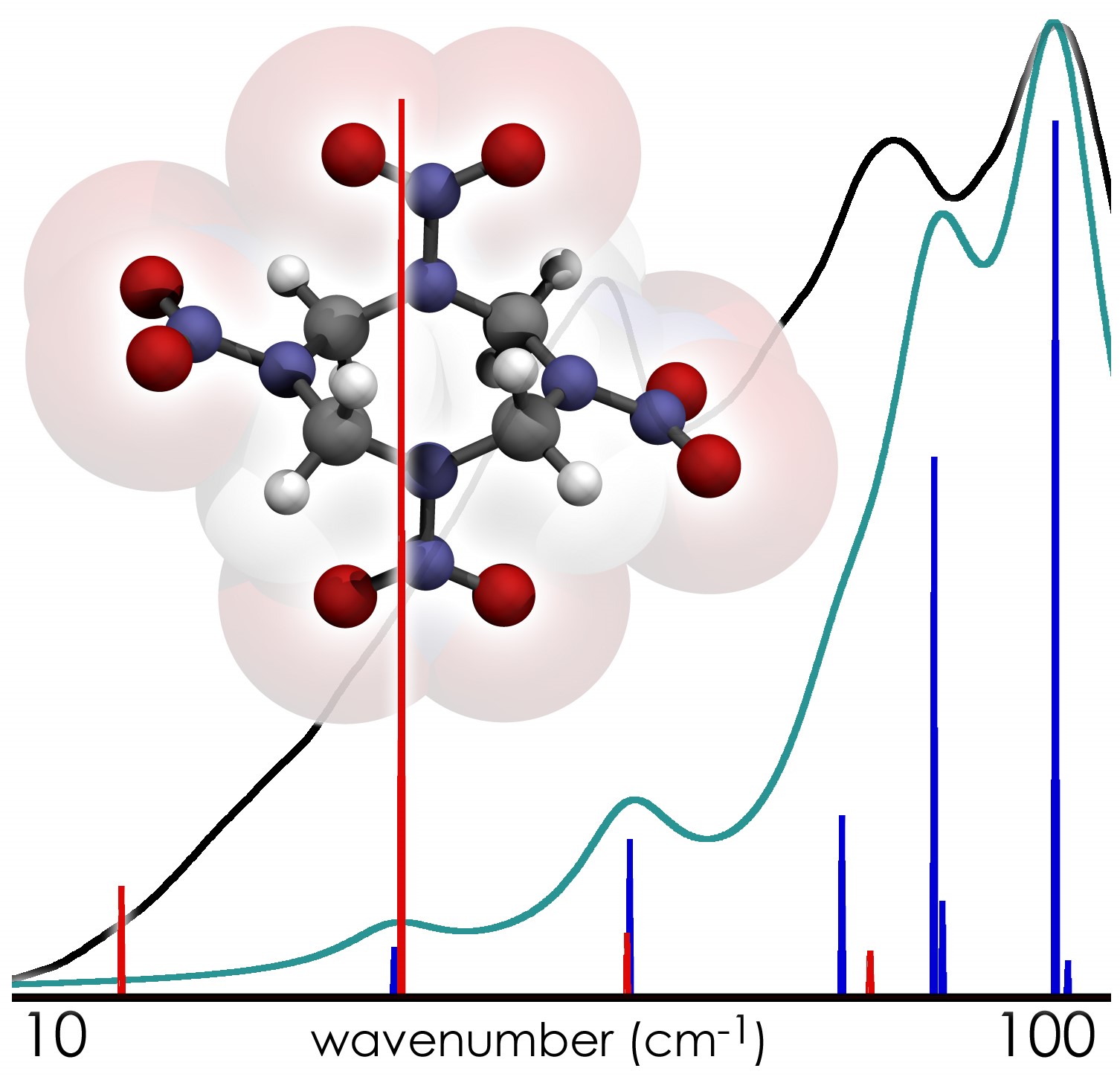
Their first solid-state computational study, involving the simulation of the THz spectrum of crystalline HMX, revealed that this spectrum contained features which could not be reproduced by the study of the individual HMX molecules alone. Prominent features in the calculated solid-state spectrum were found to arise entirely from the relative motions of molecules in the solid, in excellent agreement with experimental data. This highly-cited study laid the groundwork for future solid-state THz simulations, but also showed the limits of the state-of-the-art in computer hardware for providing researchers a predictive tool in the study of solid-state intermolecular interactions. Simply put, these calculations could only be performed on small chemical systems because the necessary theoretical accuracy to interpret these spectra could only be achieved after many weeks of dedicated compute time for a single molecular crystal.
While machine speeds and new algorithms have incrementally reduced the time needed for researchers to study the behavior of molecular solids by quantum chemical methods, the availability of highly parallelized virtual environments like OrangeGrid have dramatically improved the ability of researchers like the Korter Group to interpret the spectra of laboratory measurements. The availability of multiple and simultaneous virtual machine instances has greatly expanded the study of molecular systems as well as the study of the theoretical methods themselves. Instead of waiting for a survey of different theoretical methods to be completed in series, these same surveys can now be done in parallel to explore the limits of modern quantum chemical methods and their applications to aspects of chemical behavior not otherwise practical with commodity hardware.
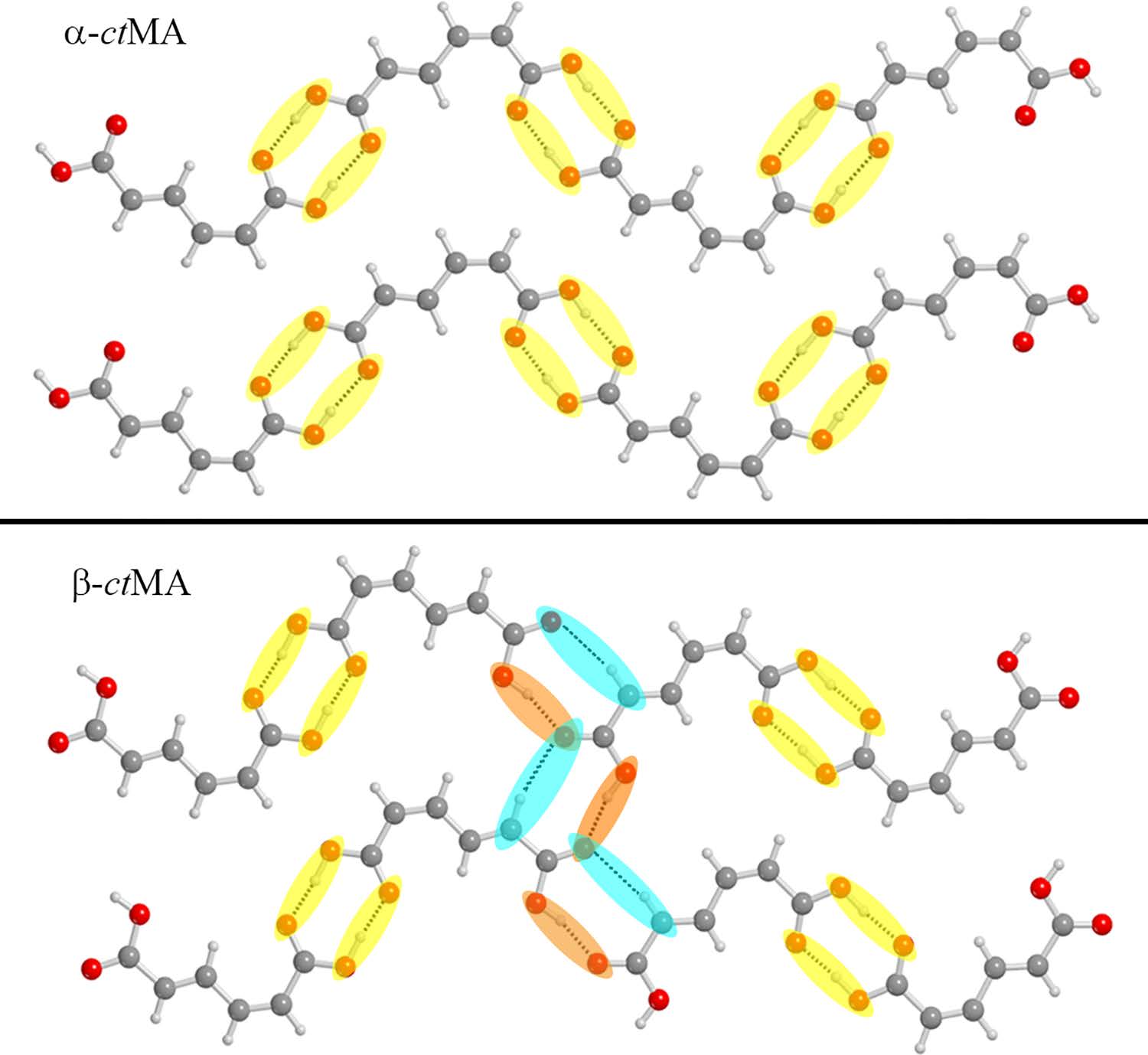
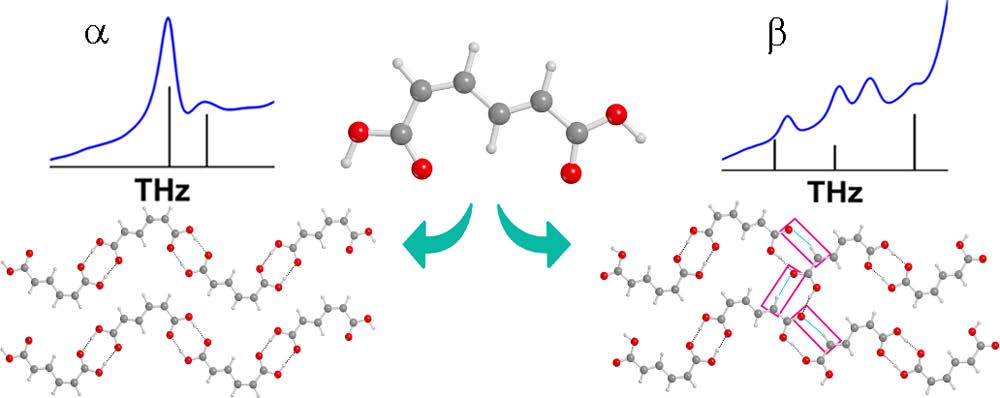
Recently published work by the Korter Group has capitalized on the wealth of data from past methodological surveys to focus greater attention on subtler phenomena within molecular crystals – crystal polymorphism and co-crystal interactions. Polymorphism, the ability of the same molecule to produce more than one crystal form, is often best characterized by the differences in the interactions between neighboring molecules in the crystal cell – differences for which THz spectroscopy is ideally suited (Crystal Growth & Design). Terahertz spectroscopy is also an excellent tool for studying the differences in structure and interaction energies among molecules whose crystals contain another bound molecule, or co-crystal. The combination of THz experiments with simulated spectra and relative energy calculations performed with OrangeGrid have been used to understand the properties of the sweetener D-sorbitol and its differences in behavior upon hydration with the sugar mannitol (Journal of Physical Chemistry A).
Funding: National Science Foundation (CHE-1301068)
Recent Papers Supported By OrangeGrid:
* A. J. Zaczek and T. M. Korter, Cryst. Growth Des. 2017, 17, 4458−4466
* T. M. Dierks and T M. Korter, J. Phys. Chem. A 2017, 121, 5720−5727